Abstract
Cardiovascular diseases (CVDs) are the major causes of mortality and morbidity worldwide as well as in the Indian subcontinent, causing more than 25% of deaths. It has been predicted that these diseases will increase rapidly in India, making it a host to more than half the cases of heart disease in the world within the next 15 years. The World Health Organization (WHO) reports that in the year 2005 CVDs caused 17.5 million (30%) of the 58 million deaths that occurred worldwide. In the recent times, the association of metabolic syndrome (MS) is strongly linked with CVDs. MS is defined as a constellation of metabolic disorders in an individual. The main components of MS are dyslipidemia (higher triglyceride, low-density lipoproteins [LDL] and low high-density lipoproteins [HDL]), elevated blood pressure (BP), dysregulated glucose homeostasis, abdominal obesity and insulin resistance. Being one of the most widespread diseases in the world, almost half of the population of specific age groups in developed countries is affected by it. Studies have shown that the independent risk factors associated with MS increase the likelihood of CVDs. It has been postulated that excess intake of fructose promotes cell dysfunction, inflammation, intra-abdominal (visceral) adiposity, atherogenic dyslipidemia, weight gain, insulin resistance, hypertension thereby aggravating the chances for developing MS, type 2 diabetes and coronary heart disease.
Keywords: Cardiovascular diseases, metabolic syndrome, dyslipidemia, abdominal obesity, fructose, insulin resistance
Introduction
As the engines of health transition gather pace, the epidemic of cardiovascular diseases (CVDs) is accelerating globally, advancing across regions and social classes. CVDs, one of the noncommunicable diseases have become the major public health problem in developed and developing countries. Globally CVD deaths represent about 30% of all deaths. As per the World Health Organization (WHO) reports, it is predicted that almost 23.6 million people will die from CVDs, mainly from heart disease and stroke by 2030, both becoming the single leading cause of death. Different studies are throwing light on the diseases associated with increased CVD risk such as metabolic syndrome (MS) as with lifestyle changes new and more complex disease conditions have emerged. The new millennium is witnessing the emergence of a modern epidemic, i.e., MS, with frightful consequences to the health of humans worldwide and its associated comorbid conditions. Studies have shown that MS patients possess a significantly greater risk for the development of CVD in general and coronary artery disease (CAD) in particular, studies have even reported a positive correlation between MS and carotid atherosclerosis.
The etiology of CVD in patients with MS may involve: coronary atherosclerotic disease, arterial hypertension, left ventricular (LV) hypertrophy, diastolic dysfunction, endothelial dysfunction, coronary microvascular disease and autonomic dysfunction.
The pathogenesis of CVD in the MS is multifactorial as it can be caused by one or more factors associated with this condition such as the systemic abnormalities, insulin resistance, diabetes and/or inflammation. It is seen that each component of MS independently affects cardiac structure and function, but their combination under this syndrome seems to carry additional risk.
MS is a complex disease bearing a high socioeconomic cost and being considered as a major epidemic worldwide. Although many definitions and classifications of MS are available two definitions in the widespread are used globally; one proposed by the WHO and other by the National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP III) (Table 1).
Broadly MS is defined as concurrence of overweight, abdominal fat distribution, dyslipidemia, disturbed glucose and insulin metabolism and hypertension (Fig. 1), historically the concept of MS dates back to 1988 when Reaven for the first time had put forward the concept of syndrome X, which was later named as MetS. However in 1999, WHO introduced the term ‘metabolic syndrome’ to include the cluster of factors as a clinical entity. Recent studies have also added other abnormalities such as chronic pro-inflammatory, hyperuricemia, prothrombogenic states, nonalcoholic fatty liver disease (NAFLD) and sleep apnea to the entity of this syndrome making its definition even more complex. Along with being a risk factor for CVDs, MS even predisposes an individual
|
Table 1. Consensus Definitions from Different Associations on Metabolic Syndrome
|
|
National Cholesterol Education Program-Adult Treatment Panel III, 2001
|
American Heart Association/National Heart, Lung and Blood Institute Scientific Statement, 2005
|
International Diabetes Federation, 2006
|
Harmonizing the Metabolic Syndrome, 2009
|
|
Three or more of the following:
|
Measure (any 3 of 5 constitute diagnosis of metabolic syndrome)
|
Central obesity as defined by ethnic/racial, specific WC and two of the following:
|
Three or more of the following:
|
|
WC >102 cm for men, >88 cm for women
|
WC >102 cm in men, >88 cm in women
|
Triglycerides ≥150 mg/dL
|
Central obesity as defined by ethnic/racial, Specific WC
|
|
Triglycerides ≥150 mg/dL
|
Triglycerides ≥150 mg/dL or on drug treatment for elevated triglycerides
|
HDL-chol <40 mg/dL for men; <50 mg/dL for women
|
Triglycerides ≥150 mg/dL or on drug treatment for elevated triglycerides
|
|
HDL-chol <40 mg/dL in men; <50 mg/dL in women
|
HDL-chol <40 mg/dL in men; <50 mg/dL in women or on drug treatment for reduced HDL-chol
|
BP ≥130/85 mmHg
|
HDL-chol <40 mg/dL in men; <50 mg/dL in women or on drug treatment for reduced HDL-chol
|
|
BP ≥130/85 mmHg
|
BP ≥130/85 mmHg or on antihypertensive drug treatment in a patient with a history of hypertension
|
FPG ≥100 mg/dL
|
BP ≥130/85 mmHg or antihypertensive drug treatment
|
|
FPG ≥110 mg/dL
|
FPG ≥100 mg/dL or on drug treatment for elevated glucose
|
|
FPG ≥100 mg/dL or on drug treatment for elevated glucose
|
WHR = Waist-to-hip ratio; WC = Waist circumference; BP = Blood pressure; FPG = Fasting plasma glucose; Chol = Cholesterol.
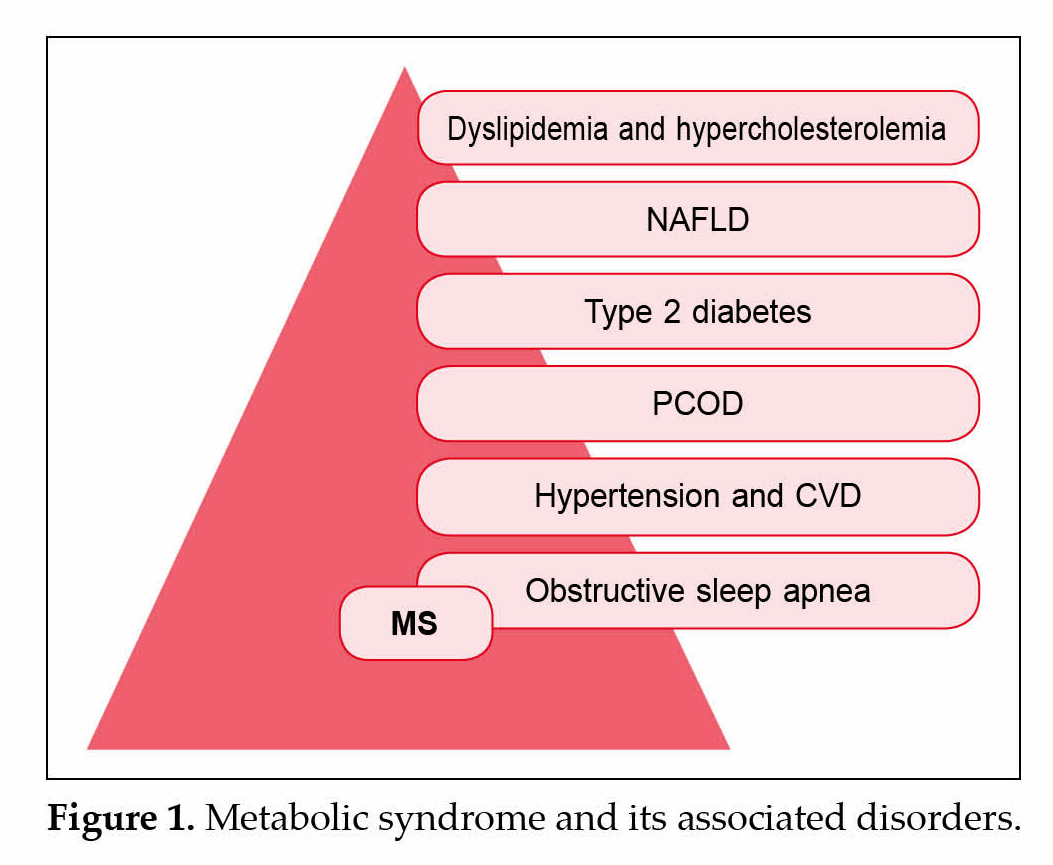
to a greater risk for developing type 2 diabetes. Changes in human behavior, high energy fast food environment, sedentary lifestyle, have recently been attributed to be associated with progression towards MS.
In the present world, there has been an augmented understanding of MS and its associated diseases followed by a subsequent increase in clinical attention directed towards its prevention, due to its strong association with premature morbidity and mortality. Numerous studies have reached to the consensus now that insulin resistance and obesity are main determining factors involved in the common pathologic mechanism of the MS and its associated comorbid conditions. Evidences have suggested that the progression towards MS begins early in life and with persistence from childhood to adolescent/adult life results in type 2 diabetes, CVDs and other associated diseases.
The symptoms of MS develop over a predisposed background thought to be established at a young age and are not necessarily manifestations of age, recent trends in modern diets, habits, lifestyle changes likely influencing health and behavior in increasingly younger populations makes up for a dangerous predisposition.
Pathophysiology of Metabolic Syndrome and CVD Progression
Understanding the pathophysiology responsible for MS will prove to be a beneficial aid for best treatment options. While the exact mechanisms responsible for increased CVD risk have not been elucidated, studies have thrown light on insulin’s action and the role of
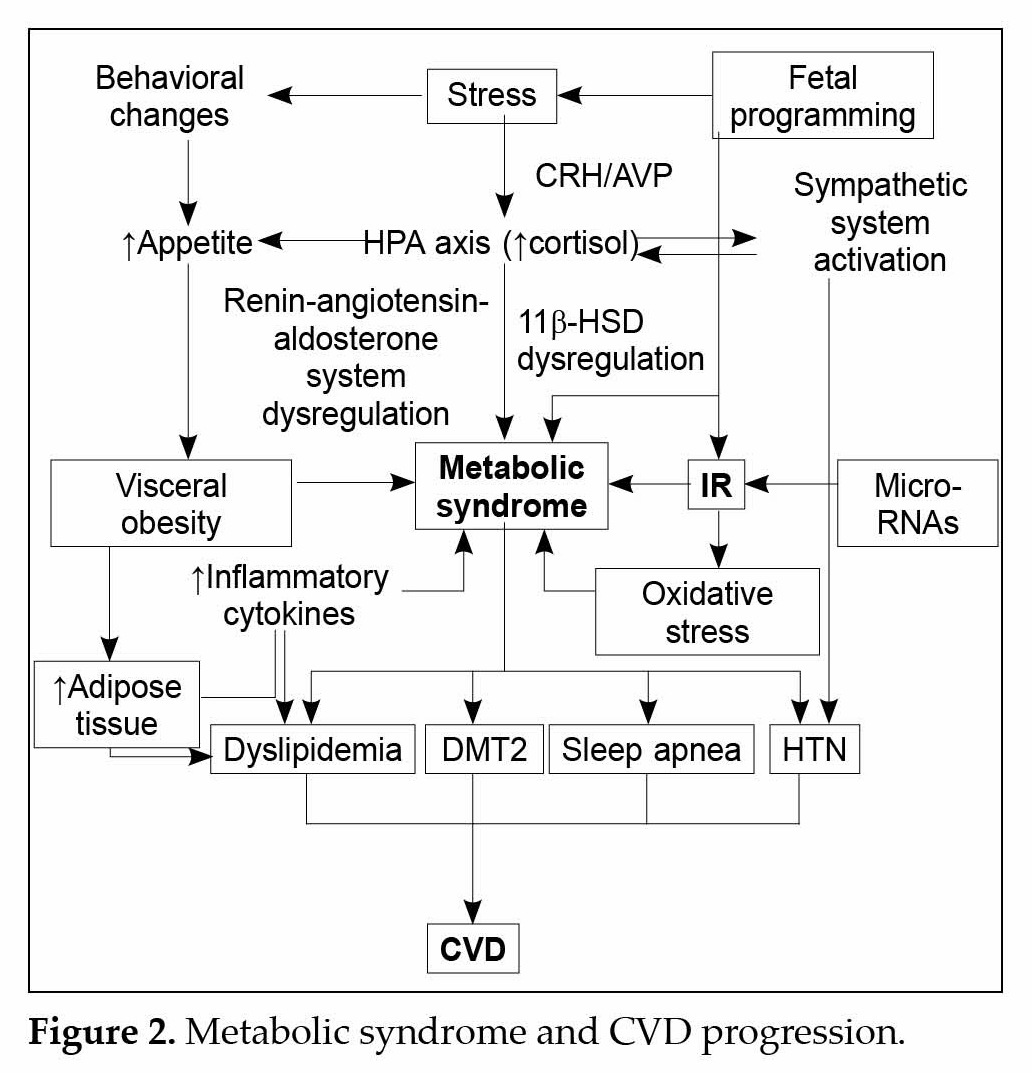
obesity and its associated pathological mechanisms (Fig. 2). The excess adiposity associated with MS plays an important role towards the progression of MS associated CVD. Obesity especially abdomino-visceral, is associated with certain pathogenic factors that contribute to normal glucose homeostasis: high plasma levels of free-fatty acids (FFAs), increased hepatic glycogenesis and peripheral insulin resistance. It is seen that in obesity, cytokines mediated release of inflammatory molecules such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, plasminogen activator inhibitor (PAI), C-reactive protein and resist in occurs from adipose tissues and immune cells due to the initiation of a chronic inflammatory state via cytokines. The link between obesity and inflammation stems from the fact that pro-inflammatory cytokines are over expressed in obesity, this inflammatory process acts as a homeostatic mechanism to prevent the accumulation of excess fat. It is established that the starting signal for inflammation in obesity is overfeeding and the pathway origins in all metabolic cells e.g., in adipocyte, hepatocyte or myocyte.
Clinical and non-clinical studies have shown that consumption of nutrients may acutely evoke inflammatory responses, furthermore metabolic cells such as adipocytes respond to this insult by beginning inflammatory response. It is seen that lipid storage and weight increase requires an anabolic process while inflammation stimulates catabolism including lipolysis, as a result due to chronic lipolysis FFAs are liberated continuously and are further transferred via portal vein to liver. Studies have shown that increased plasma FFAs along with inflammatory cytokines trigger a response that results in decreased insulin sensitivity in tissues that depend on insulin which is caused by inhibition of receptor signaling, this situation is also referred to as insulin resistance (IR) further leading to increased insulin synthesis and secretion by β pancreatic cells and resulting in compensatory hyperinsulinemia, following it FFAs are oxidized simultaneously in the liver, triggering neoglucogenesis and thus increasing glycemia. Concomitantly, there is an increase in the synthesis of very low-density lipoproteins (VLDLs) that further generate small, dense atherogenic LDLs.
Studies have shown an extensive role of TNF-α in the systemic inflammatory response triggered by obesity, it is seen that within adipose tissue macrophages account for maximum TNF-α production and it has also been seen that TNF-α expression levels are higher in obese patients, a link between increased levels of circulating TNF-α levels and IR has been established. The pathway of IR via TNF-α occurs through serine phosphorylation (inactivation) of both the insulin receptor and insulin receptor substrate-1 (IRS-1), as a result a diminished activation of phosphoinositide 3-kinase (PI3K) occurs which is a main governing molecule of insulin’s metabolic effects. One of the mechanisms by which TNF-α is thought to trigger IR is via activation of nuclear factor-κB (NF-κB) signaling, which further results in activation of inflammatory cascade. Another mechanism by which TNF-α is thought to contribute to IR is through the elevated levels of circulating FFAs caused by induction of lipolysis and stimulation of hepatic lipolysis; however, this mechanism has only preliminary supporting evidence and extensive studies are needed to completely validate the findings.
Studies have depicted that insulin’s action also plays a crucial role towards CVD progression in MS patients. Deedwania in his study; noted that insulin’s action can lead to hypertension via stimulation of vascular smooth muscle cell hypertrophy; in addition, insulin could also cause hypertriglyceridemia and high-density lipoprotein (HDL) cholesterol through increased catecholamines. It is also reported that insulin can lead to secretion of prothrombogenic PAI-1.
Studies have shown that hyperinsulinemia may even lead to increased sensitivity to angiotensin II, which further could result in increases in cell growth, PAI-1, intracellular adhesion molecule-1, etc. Defects in insulin sensitivity may interfere with insulin-stimulated vasodilation. IR is also associated with endothelial dysfunction, which is characterized by impaired endothelium-dependent vasodilation, reduced arterial compliance and accelerated process of atherosclerosis. Along with obesity and IR, studies have even thrown light on the role of matrix metalloproteinase (MMPs) in MS and associated CVDs. Progression towards MS associated CVD begins via alterations of the arterial vasculature, which begins with endothelial dysfunction and lead to micro- and macrovascular complications. It is seen that remodeling of the endothelial basal membrane that promotes erosion and thrombosis occurs due to multifactorial pathogenesis that includes leukocyte activation, increased oxidative stress and also an altered MMP. Being endopeptidases, the primary role of MMPs is to degrade matrix proteins, such as collagen, gelatins, fibronectin and lamin, and can be secreted by several cells within vascular wall. The activity of MMPs is regulated by tissue inhibitors of MMP (TIMPs) and also by other molecules, such as plasmin. The role of MMPs in plaque instability causing serious vascular complications has been reported in several studies. It has been demonstrated that an impaired MMP or TIMP expression is associated with higher risk of all-cause mortality.
In the recent years, MMPs have garnered considerable interest due of their association with many disease conditions. It is seen that different components of the MS provide an impetus for MMP synthesis and even their activity; these include hypertension, dyslipidemia, hyperglycemia, pro-inflammatory and pro-oxidant markers, on the other hand, anti-inflammatory cytokines like adiponectin are inversely associated with MMPs. Extensive studies have come up to the conclusion that among the several MMPs collagenases (MMP-1 and MMP-8) and gelatinases (MMP-2 and MMP-9) are strongly associated with MS progression and its associated diseases, even few studies targeting MMPs in patients coronary diseases and diabetes and have shown fruitful results. In the near future, targeting MMPs and their activators can prove beneficial in treating and understanding the MS complexity and its associated diseases.
Nutritional Factors Influencing Metabolic Syndrome
In the recent times, a trend towards the shift in the energy balance accompanied by sedentary lifestyle and increased caloric intake is gathering considerable importance, and is being attributed to technological advances and improved economic status in Western countries and even developing countries. Studies have shown that the Westernization of diets, along with high calorie foods is certainly becoming an important contributor to MS epidemic, and the increased incidence of the MS now even threatens developing countries.
In the past, physicians and scientists have made an association between dietary energy from fat and body fat, following which a large market is being popularized and promoted for low fat diets, interestingly however, the decline in dietary fat consumption has not corresponded to a decrease in obesity in fact, an opposite trend has emerged. It is seen that diets high in saturated fats induce weight gain, IR and hyperlipidemia in humans and animals. Despite putting effortless emphasis on fat reductions no significant benefits relative to the obesity epidemic have emerged, increasing evidence now suggests that the rise in consumption of carbohydrates, particularly refined sugars high in fructose, appears to be at least one very important contributing factor. Recent epidemiological and biochemical studies clearly suggest that high-fructose intake may play an important role in progression towards MS.
At present, the market is flooded with large quantities of popular, convenient, prepackaged foods, soft drinks and juice beverages containing sucrose or high-fructose corn syrup. Fructose, which is found naturally in many fruits, is now consumed by humans in large quantities in the commonly available popular foods. Studies have shown that an approximate 25% increase in per capita fructose consumption over the past 30 years clearly co-exists, which increase in the prevalence of obesity and MS; high-fructose diets have been shown to induce IR, weight gain, hyperlipidemia and hypertension in several animal models including rats, hamsters and dogs.
In human studies, fructose consumption is seen to be associated with the development of hepatic and adipose tissue IR and dyslipidemia due to its ability to induce hepatic de novo lipogenesis (Fig. 3). Different biomechanical studies have suggested that sugar consumption causes adverse effects because of rapid hepatic metabolism of fructose which is catalyzed by fructokinase C, which further results in increased uric acid levels and even generates substrate for de novo lipogenesis.
Studies have shown glucose transporter 5 (GLUT5) present at the brush border and basolateral membranes of the jejunum aids in the absorption of fructose from the intestine into the portal blood; as a result massive fructose uptake by the liver occurs via this route. It is noted that the hepatic metabolisms of both glucose and fructose are different; fructose is phosphorylated by fructokinase, forming fructose-1-phosphate, which can then be converted to several three-carbon molecules, including glyceraldehyde, dihydroxyacetone phosphate and glyceraldehyde 3-phosphate. It is seen that some of these three-carbon molecules via the process of gluconeogenesis could be converted to glucose, or could also be used to generate other products such as triglycerides (TGs), which further can be packaged into VLDL by the liver. As VLDLs travel through the bloodstream, TGs can be hydrolyzed by lipoprotein lipase to form nonesterified fatty acids (NEFAs) and monoacylglycerol, which are further taken up by adipose tissue and resynthesize to TGs, therefore excessive fructose consumption can lead to high levels of FFAs and obesity. It is already stated that the role of the adipose tissue is to take up FFAs and store it in the form of TGs, however in obesity it is seen that this storage capacity reaches to its maximum resulting in an impaired ability of adipose tissue to acquire dietary fatty acids, as a result increased levels of fatty acids occurs in circulation.
Studies have shown that signaling abnormalities in adipocytes can also trigger lipolysis of TG stores resulting in efflux of fatty acids into the bloodstream thereby augmenting the problem. Few studies have thrown light that high levels of NEFAs in the bloodstream have a positive correlation between obesity, IR, type 2 diabetes and metabolic dyslipidemia, these NEFAs are eventually taken up ectopically by nonadipose tissues such as the liver and skeletal muscle, where they may be stored as TG or diacylglycerol and interfere with metabolic pathways such as the response to insulin, contributing to IR and MS. Numerous studies have shown that the build-up of lipids in the liver and other tissues in obesity contributes to an increased mitochondrial oxidation of fatty acids, further generating peroxidation products that stimulates IκB kinase (IKK)β and, therefore, NF-κB activation. Various studies have even found a correlation between fatty acid or lipid treatment and NF-κB activation. Under
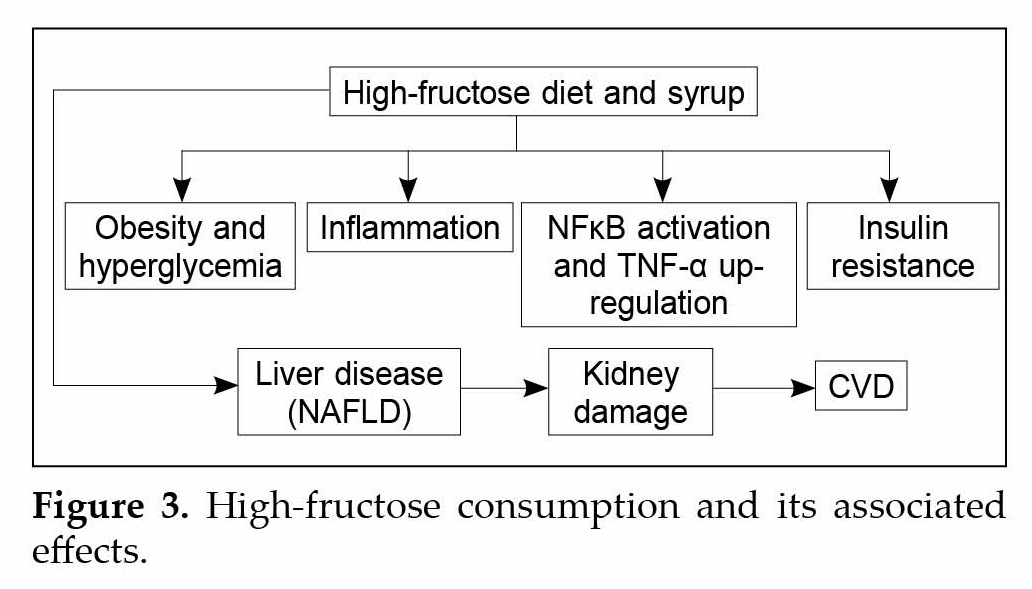
basal conditions NF-κB is found in the cytosol bound to its inhibitor, IkB, but upon activation of IKKβ, which phosphorylates IkB and marks it for degradation, NF-κB is allowed to enter the nucleus, where it induces transcription of specific genes.
The proteins encoded by these genes include pro-inflammatory cytokines such as PAI-1, TNF-α, IL-6 and IL-1β; however, the mechanisms responsible for IKKβ and NF-κB activation in obesity are unclear. TNF-α being a mediator of inflammation and immune response is a versatile cytokine that alters tissue remodeling, epithelial cell barrier permeability, activation of macrophages and recruitment of inflammatory filtrates different downstream signaling cascades activated by TNF-α have been elucidated. TNF-α, by binding to various receptors such as TNFR1 results in the activation of various transcription factors e.g., NF-κB as well as intrinsic and extrinsic apoptotic cascades mediated by caspase-8 and cytochrome C.
Recent studies have shown the involvement of TNF-α in mitochondrial membrane destabilization, resulting in formation of pathological pores causing mitochondrial permeability transition thereby activating the intrinsic pathway of apoptosis mediated by cytochrome C in many diseases. Numerous nonclinical studies have delineated the effect of fructose and its associated activated pathological pathways on the progression towards MS associated CVDs in rodents. In a study,
Shiu et al delineated the apoptotic and antisurvival effects on rats hearts when administered high fructose diet (HFD). It was seen that rats on HFD besides having elevated levels of all MS markers, had abnormal myocardial architecture, enlarged interstitial space and increased cardiac apoptotic cells. The role of intrinsic and extrinsic apoptotic markers such as Fas-dependent apoptotic proteins (TNF-α, TNFR1, Fas ligand, Fas receptor, FADD, activated caspase-8 and activated caspase-3), mitochondria dependent apoptotic proteins (Bax, Bak, Bax/Bcl-2, Bak/Bcl-xL, cytosolic cytochrome C, activated caspase-9 and activated caspase-3) was delineated in the study. Rats on HFD had up-regulated levels of the above stated markers. Further cardiac insulin-like growth factor 1 (IGF-1-related survival proteins (IGF-1, IGF-1R, p-PI3K and p-Akt) and Bcl-2 family associated pro-survival proteins (Bcl-2 and Bcl-xL) were down-regulated in rats on HFD.
Parks et al and Katan et al, showed in their short-term studies that diets rich in carbohydrates, particularly sugars (sucrose, fructose) resulted in increase in serum triacylglycerol concentrations and decreased HDL concentration, therefore indicating a risk towards developing CVD. Few studies have also thrown light on the involvement of various oxidative stress markers (nicotinamide adenine dinucleotide phosphate or NADPH) and pro-inflammatory cytokines (IL-1, IL-6) and hypothesize a possible role of NF-κB and TNF-α in the progression of MS thereby leading to CVDs. Numerous studies have found that dietary composition of carbohydrate can result in development of left ventricular hypertrophy and cardiac pathology. It is believed that with increased concentrations of fructose diets the trend towards CVD risk will markedly rise in near future.
Conclusion
With rising financial implications and with a concomitant impact on human health, MS in the recent past has gathered considerable concern; its presence is an important risk factor for the development of CVDs and type 2 diabetes. At present, the key principles involved in the management of patients with MS are early identification of patients, effective treatment regular follow-up, pharmacological therapy and lifestyle modifications. In the current scenario, the mechanisms that contribute to MS associated diseases remain unclear, extensive research is underway that might help in understanding the pathological pathways and novel treatment options. The most important contributory factors which have emerged as the important links in MS are sedentary lifestyle, altered dietary requirements and obesity.
The consumption of fructose has increased, largely because of an increased consumption of soft drinks and many juice beverages containing sucrose or high-fructose corn syrup. Dietary high-fructose intake has been suggested to be an important factor contributing to the development of symptoms of MS. Recent evidence suggests that fructose feeding in rats develops the features of the MS model in many of the same pathophysiological deficits as noted in MS in humans, such as IR, dyslipidemia, hyperinsulinemia, hypertricylglycerolemia, impaired glucose tolerance, increased uric acid levels, hypertension, myocardial functional abnormalities and heart failure. If not alarmed and the different economies do not make necessary interventions to the growing MS epidemic, individuals from all age groups will be severely affected limiting their full overall development and progression.
Suggested Reading
- Gaziano TA, Bitton A, Anand S, Abrahams-Gessel S, Murphy A. Growing epidemic of coronary heart disease in low- and middle-income countries. Curr Probl Cardiol. 2010;35(2):72-115.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. 1997;349(9063):1436-42.
- World Health Organization ((WHO). Cardiovascular disease. Fact sheet N°317. Geneva. September 2009. Available at: http://wwwwhoint/mediacentre/factsheets/fs317/en/indexhtml
- Hutcheson R, Rocic P. The metabolic syndrome, oxidative stress, environment, and cardiovascular disease: the great exploration. Exp Diab Res. 2012;2012: 271028.
- Iglseder B, Cip P, Malaimare L, Ladurner G, Paulweber B. The metabolic syndrome is a stronger risk factor for early carotid atherosclerosis in women than in men. Stroke. 2005;36(6):1212-7.
- Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics - 2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18-e209.
- Kim JY, Mun HS, Lee BK, Yoon SB, Choi EY, Min PK, et al. Impact of metabolic syndrome and its individual components on the presence and severity of angiographic coronary artery disease. Yonsei Med J. 2010;51(5):676-82.
- Schernthaner G. Cardiovascular mortality and morbidity in type-2 diabetes mellitus. Diabetes Res Clin Pract. 1996;31 Suppl:S3-13.
- Lakka HM, Laaksonen DE, Lakka TA, Niskanen LK, Kumpusalo E, Tuomilehto J, et al. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA. 2002;288(21):2709-16.
- Ilkun O, Boudina S. Cardiac dysfunction and oxidative stress in the metabolic syndrome: an update on antioxidant therapies. Curr Pharm Des. 2013;19(27):4806-17.
- Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, et al; American Heart Association; National Heart, Lung, and Blood Institute. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735-52.
- Grundy SM. Metabolic syndrome: a multiplex cardiovascular risk factor. J Clin Endocrinol Metab. 2007;92(2):399-404.
- Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond). 2008;114(3):195-210.
- Nicolson GL. Metabolic syndrome and mitochondrial function: molecular replacement and antioxidant supplements to prevent membrane peroxidation and restore mitochondrial function. J Cell Biochem. 2007;100(6):1352-69.
- Klein BE, Klein R, Lee KE. Components of the metabolic syndrome and risk of cardiovascular disease and diabetes in Beaver Dam. Diabetes Care. 2002;25(10):1790-4.
- Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162.
- Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15(7):539-53.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486-97.
- Tuomilehto J. Cardiovascular risk: prevention and treatment of the metabolic syndrome. Diabetes Res Clin Pract. 2005;68 Suppl 2:S28-35.
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595-607.
- Consultation W. Definition, diagnosis and classification of diabetes mellitus and its complications: Part; 1999.
- Kassi E, Pervanidou P, Kaltsas G, Chrousos G. Metabolic syndrome: definitions and controversies. BMC Med. 2011;9:48.
- Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. 2009;2(5-6):231-7.
- Oda E. Metabolic syndrome: its history, mechanisms, and limitations. Acta Diabetol. 2012;49(2):89-95.
- Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med. 2005;56:45-62.
- Tappy L, Lê KA, Tran C, Paquot N. Fructose and metabolic diseases: new findings, new questions. Nutrition. 2010;26(11-12):1044-9.
- Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond). 2005;2(1):5.